Definition
Pheochromocytoma is a rare tumor typically found in the adrenal glands, which are situated above each kidney. These glands are integral to the body's endocrine system and are responsible for hormone production.
Usually benign (non-cancerous), a pheochromocytoma primarily develops in one adrenal gland, though tumors can occasionally occur in both glands.
Causes
The exact cause of pheochromocytoma remains uncertain. Tumors form in chromaffin cells situated at the center of the adrenal glands. These cells release hormones, particularly adrenaline (epinephrine) and noradrenaline (norepinephrine), which regulate various bodily functions such as heart rate, blood pressure, and blood sugar levels.
Adrenaline and noradrenaline prompt increases in blood pressure and heart rate. Pheochromocytoma leads to an excessive release of these hormones even when they're not required.
While most chromaffin cells reside in the adrenal glands, some clusters are also found in other areas like the heart, head, neck, bladder, abdominal wall, and along the spine. Tumors arising from chromaffin cells outside the adrenal glands are termed paragangliomas. These tumors can induce similar physiological effects on the body as pheochromocytoma.
Risk factor
While most pheochromocytomas are detected in individuals aged 20 to 50, these tumors can develop at any age.
Individuals with certain rare hereditary conditions face an elevated risk of developing pheochromocytoma or paraganglioma. Tumors associated with these disorders are more likely to be malignant and may occur in both adrenal glands. These genetic conditions include:
- Multiple endocrine neoplasia type 2 triggers tumors across various parts of the endocrine system. Both type 2A and type 2B are linked with pheochromocytoma. Additionally, tumors associated with this condition can manifest in other regions of the body, such as the thyroid, parathyroid, lips, tongue, and digestive system.
- Von Hippel-Lindau disease can lead to tumors in numerous locations, including the central nervous system, endocrine system, pancreas, and kidneys.
- Neurofibromatosis type 1 is characterized by multiple skin tumors (neurofibromas), skin pigmentation spots, and tumors on the optic nerves.
- Hereditary paraganglioma syndrome a congenital disorder predisposing individuals to pheochromocytoma or paraganglioma.
Symptoms
The signs and symptoms of pheochromocytoma commonly include:
- High blood pressure
- Headaches
- Excessive sweating
- Rapid heartbeat
- Tremors
- Paleness
- Shortness of breath
- Symptoms resembling panic attacks
Less frequently observed signs and symptoms may include
- Anxiety
- Blurred vision
- Constipation
- Weight loss
Symptoms of pheochromocytoma can be persistent or may occur or worsen intermittently, known as spells. These spells can be triggered by specific activities or conditions, consumption of foods high in tyramine (a substance affecting blood pressure), and certain medications.
Activities or conditions that can exacerbate symptoms include:
- Strenuous physical activity
- Anxiety or stress
- Changes in body position (e.g., from sitting to lying down or standing)
- Childbirth
- Surgery and medications inducing anesthesia
Tyramine-rich foods, which affect blood pressure, can also worsen symptoms. Examples include:
- Cheese
- Beer and wine
- Chocolate
- Cured or smoked meats
Certain medications that may exacerbate symptoms include:
- Monoamine oxidase inhibitors (MAOIs), such as phenelzine, tranylcypromine, and isocarboxazid
- Stimulants like amphetamines or cocaine
Diagnosis
To diagnose pheochromocytoma, your doctor will likely conduct several tests:
Laboratory Tests: These tests measure levels of adrenaline, noradrenaline, or their metabolites in your body. They may include:
- 24-hour urine test: Collecting a urine sample every time you urinate over a 24-hour period.
- Blood tests.
Imaging Examinations: If blood tests suggest the presence of pheochromocytoma or paraganglioma, your doctor may perform one or more radiological exams to detect a possible tumor. These may include:
- CT scan
- MRI
- Metaiodobenzylguanidine (MIBG) scan: A technology that detects small amounts of injected radioactive material taken up by the tumor.
- PET scan: Detects radioactive material taken up by tumors.
Genetic Testing: Your doctor may recommend genetic testing to determine if pheochromocytoma is associated with an inherited disorder. This information is valuable because:
- Some inherited disorders can cause various medical conditions, necessitating further evaluation.
- Certain disorders are more prone to recurrence or malignancy, influencing therapy decisions or long-term monitoring plans.
- Test results may indicate the need to screen other family members for pheochromocytoma or related conditions.
Management
The primary treatment for pheochromocytoma is surgery to remove the tumor. Before the surgery, your doctor will likely prescribe specific medications to lower blood pressure.
The entire adrenal gland containing the pheochromocytoma will often be removed. However, your doctor may only remove the tumor, sparing the healthy adrenal tissue. This approach may be chosen if one adrenal gland has already been removed or if tumors are present in both glands.
If the tumor is malignant, the surgical procedure will entail removing both the tumor and any surrounding cancerous tissue.
Therapies for malignant tumors and cancer that has metastasized throughout the body may include:
- Targeted therapy involves using drugs or substances that target specific abnormalities in cancer cells, allowing them to survive.
- Chemotherapy: Utilizing potent drugs that kill rapidly dividing cancer cells.
- Radiation therapy: This treatment can alleviate symptoms of tumors that have spread to the bones and cause pain.
Complications
High blood pressure can damage organs, particularly the heart, blood vessels, brain, and kidneys. This damage can precipitate several critical conditions, including:
- Heart disease
- Stroke
- Kidney failure
- Disorders of the optic nerve
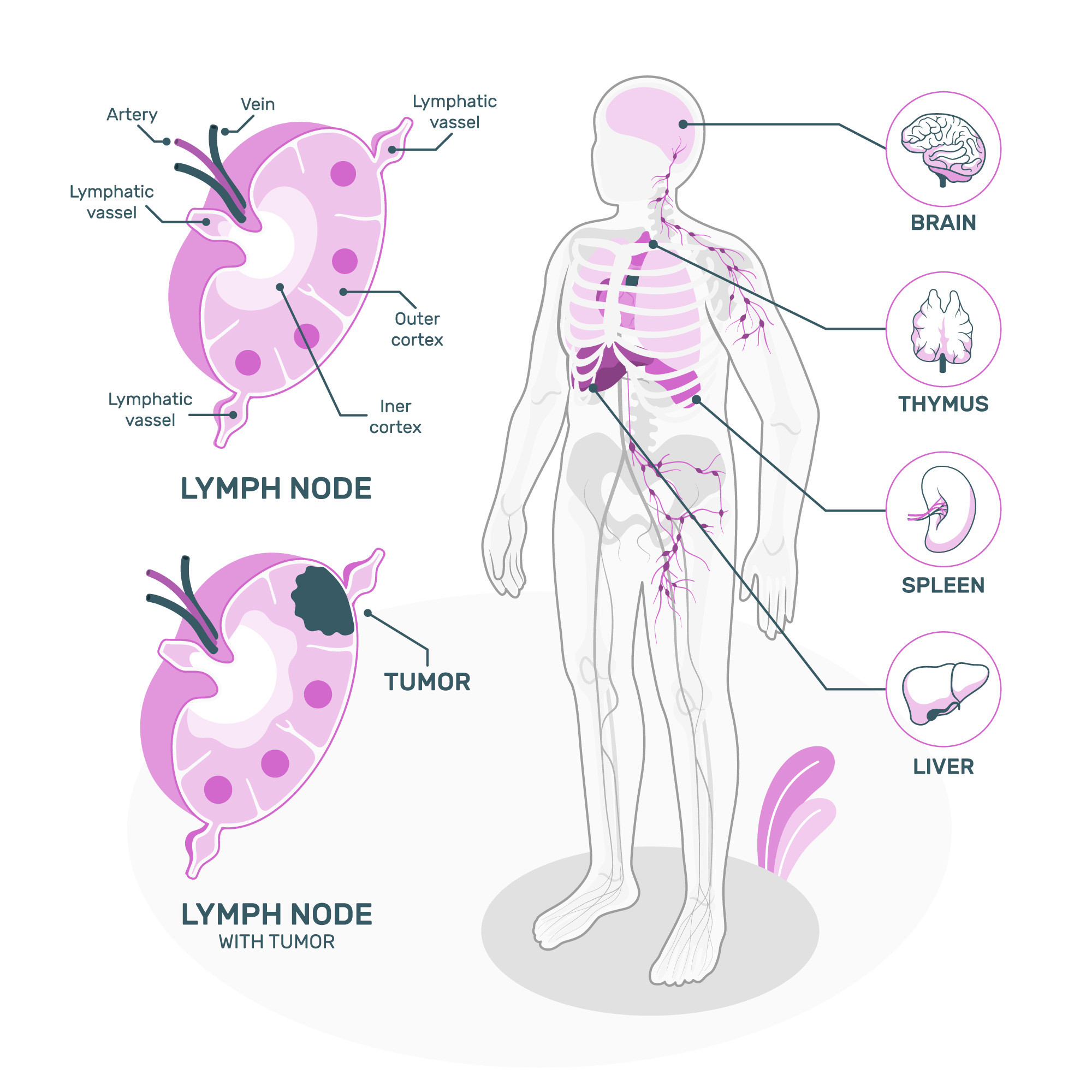
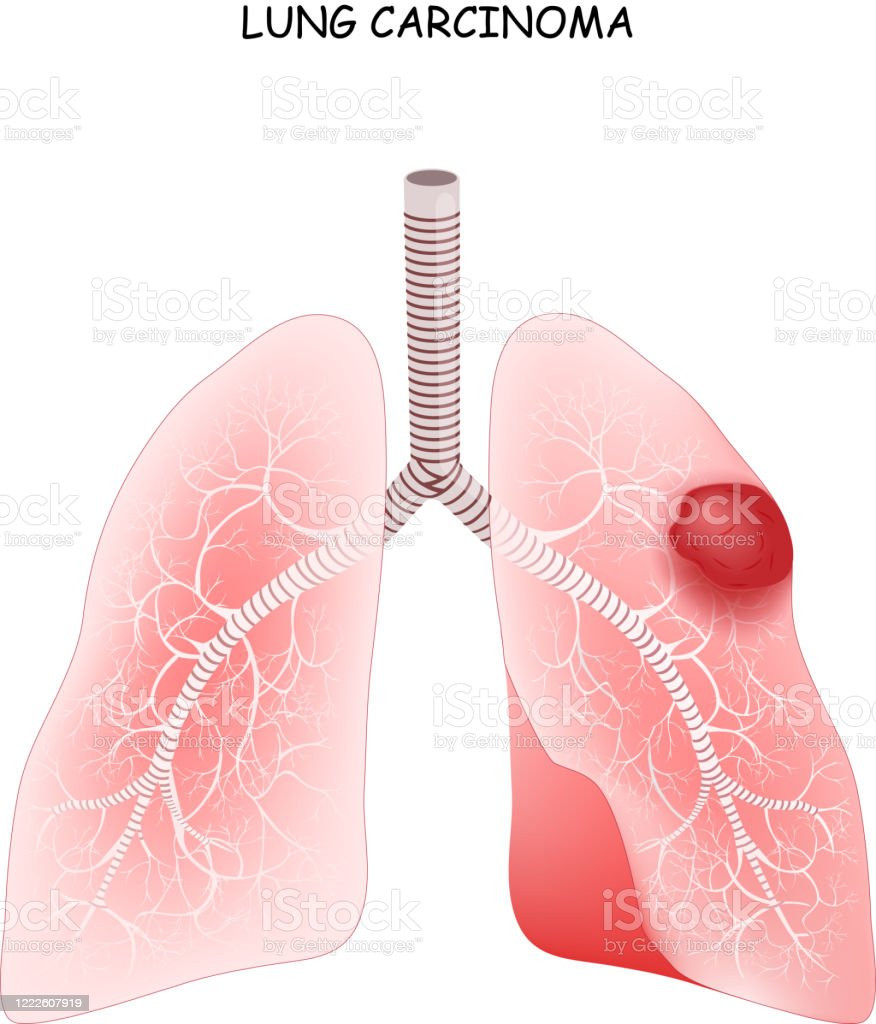
While rare, pheochromocytoma can be malignant, and cancerous cells may spread to other body parts. The most common sites for spreading cancer cells from pheochromocytoma or paraganglioma include the lymphatic system, bones, liver, or lungs.
Read more: Accounting For Acute Kidney Failure - Definition, Causes, Symptoms, and Management
Prevention
Pheochromocytoma cannot be prevented. However, if you're at risk due to inherited syndromes or genetic diseases, genetic counseling can aid in early screening for pheochromocytoma and facilitate early management.
When to see a doctor?
Schedule a consultation with a doctor if you experience any of the following:
- Difficulty controlling high blood pressure despite therapy
- Escalation of high blood pressure spells
- Family history of pheochromocytoma
- Family history of related genetic disorders such as multiple endocrine neoplasia type 2, von Hippel-Lindau disease, hereditary paraganglioma syndrome, or neurofibromatosis 1
Looking for more information about other diseases? Click here!
- dr. Monica Salim
Pheochromocytoma (2022) Mayo Clinic. Mayo Foundation for Medical Education and Research. Available at: https://www.mayoclinic.org/diseases-conditions/pheochromocytoma/symptoms-causes/syc-20355367 (Accessed: April 14, 2023).
Pheochromocytoma: Causes, symptoms & treatment (2022) Cleveland Clinic. Available at: https://my.clevelandclinic.org/health/diseases/23373-pheochromocytoma#prevention (Accessed: April 14, 2023).
Pheochromocytoma (2020) National Cancer Institute. Available at: https://www.cancer.gov/pediatric-adult-rare-tumor/rare-tumors/rare-endocrine-tumor/pheochromocytoma (Accessed: April 14, 2023).