Definition
Hemoglobinopathy is a disorder caused by variant forms of hemoglobin in red blood cells. The distribution of hemoglobin variants varies geographically worldwide, particularly in regions resembling malaria-prone areas. The presence of moderate quantities of hemoglobin variants may confer a selective advantage, providing protection against the lethal effects of malaria and enabling more individuals to reach reproductive age.
Hemoglobin C (Hb C) is relatively common among black Africans living north of the Niger River and is found in 2-3 percent of black Americans. Hemoglobin C disease occurs when the Hb C gene variant is inherited from both parents, presenting symptoms such as vague pain, jaundice, spleen enlargement, mild to moderate anemia, and occasional bleeding.
Nevertheless, individuals with this condition typically have normal lifespans, and the disease is much milder than sickle cell anemia, which is found in the same geographic range. There is a possibility that Hb C gradually replaces Hb S (the hemoglobin variant associated with sickle cell anemia) through selection processes in Africa. Unlike sickle cell anemia, Hb C does not cause early death in homozygotes (individuals with two Hb C genes), but it can offer protection against malaria.
Hemoglobin D is primarily found among individuals of Afghan, Pakistani, and Northwest Indian descent, but it also occurs in people of European descent. Hemoglobin D disease (resulting from two genes for Hb D) can cause mild hemolytic anemia. Hemoglobin E is widespread in Southeast Asia, particularly among people in Thailand, Cambodia, Laos, Malaysia, Indonesia, Vietnam, and Burma.
Hemoglobin E disease (resulting from two genes for Hb E) can cause mild microcytic anemia (characterized by small red blood cells). Hemoglobin E-thalassemia disease (resulting from one gene for Hb E and one for thalassemia) is severe and clinically very similar to major thalassemia. Hemoglobin H is most commonly found in Southeast Asia and Southern Europe and is almost always identified with thalassemia; its symptoms are similar to thalassemia.
Causes
Mutations or changes in an individual's DNA structure can cause hemoglobinopathy. The most common forms are sickle cell anemia and thalassemia. Additionally, G6PD deficiency can cause hemoglobinopathy.
Sickle Cell Anemia
Sickle cell anemia is a type of sickle cell disease that affects the shape of red blood cells, responsible for carrying oxygen throughout the body. Normally, red blood cells are round and flexible, allowing them to move easily through blood vessels. However, some red blood cells become shaped like sickles or crescent moons in sickle cell anemia. These abnormal cells also become rigid and sticky, hindering or blocking blood flow. While there is no cure for most people with sickle cell anemia, treatment can help alleviate pain and prevent complications associated with the disease. Sickle cell anemia is inherited from the sickle cell trait, which occurs when a person has one sickle cell gene and one normal gene. Individuals with only the sickle cell trait typically don't exhibit symptoms of sickle cell anemia but can pass the trait to their children. If both parents have the trait, their child may be more likely to develop sickle cell disease.
G6PD Deficiency
G6PD deficiency is an inherited condition when the body lacks sufficient amounts of an enzyme called G6PD (glucose-6-phosphate dehydrogenase). This enzyme is essential for proper red blood cell function. Insufficient levels of this enzyme can lead to hemolytic anemia, where red blood cells break down faster than they can be produced. G6PD deficiency is most commonly observed in males and is rare in females. The disorder affects approximately 10% of African American males in the US and is also prevalent in individuals from Mediterranean, African, or Asian regions. The severity of the disorder varies among different populations. The condition is typically mild in African Americans and primarily affects older red blood cells. Conversely, the disorder tends to be more severe in white populations, impacting younger red blood cells.
Thalassemia
Thalassemia is an inherited disorder characterized by abnormal production of hemoglobin, the protein responsible for oxygen transport in red blood cells. This condition results in reduced hemoglobin production and destruction of red blood cells. Thalassemia typically manifests in individuals who inherit at least two abnormal genes. Carriers who possess only one abnormal gene usually remain asymptomatic unless they carry another abnormal gene that interacts with the thalassemia gene
Sabit Cell Anemia
Cylindrical cell anemia is one of a group of congenital abnormalities known as a sickle cell disease. It affects the shape of red blood cells, which carry oxygen throughout the body. Red blood cells are usually round and flexural, so they easily move through blood vessels. In sickle cell anemia, several red blood cells are shaped like sickles or sickles. This sickle cell also becomes rigid and sticky, which can slow down or clog blood flow. There is no cure for most people with sickle cell anemia. Treatment can relieve pain and help prevent complications associated with this disease. Cylindrical cell anemia is generated from the properties of the sickle cell. The properties of the sickle cell occur when one person has a sickle cell gene and one normal gene. If you only have the properties of a sickle cell, you usually have no symptoms and have no sickle cell anemia. However, you can forward the properties of sickle cells to your children. If both parents have such properties, the child may be more likely to have some kind of sickle cell disease.
G6PD Disorders
The G6PD deficiency is a condition that is lowered. This condition occurs when the body does not have enough enzymes called G6PD (glucose-6-photo dehydrogenase). This enzyme helps red blood cells work properly. This lack of enzymes can cause hemolitic anemia. This is when red blood cells break faster than they are made. G6PD deficiency most often occurs in men and rarely occurs in women. Such disorders affect about 10% of African-American men in the US. This is also common in people from the Mediterranean region, Africa, or Asia. The severity of disorder varies, depending on the group. In Africa-American, the problem is mild. This mainly affects older red blood cells. In white people, these abnormalities are often more serious. In this group, pink blood cells are affected.
Thalassemia
Thalassemia is a congenital disorder characterized by abnormal production of hemoglobin, a protein in red blood cells carrying oxygen. They produce low hemoglobin production and destruction of red blood cells. This disease usually only occurs in people who have at least two abnormal genes. Carriers (people with only one abnormal gene) usually have no problems, unless they carry another abnormal gene interacting with thalassemia genes.
Risk factor
Risk factors for hemoglobinopathy may include:
- Family history of hemoglobinopathy
- Living in an endemic area with hemoglobinopathy diseases such as malaria or residing around the thalassemia belt
Symptoms
Symptoms of hemoglobinopathy may include:
- Increased need for sleep or excessive sleepiness
- Fatigue
- Shortness of breath
- Pain or swelling in the hands or feet
- Cold hands or feet
- Pale skin
Diagnosis
Diagnosing hemoglobinopathy typically involves a comprehensive procedure consisting of taking a medical history, performing a physical examination, and performing diagnostic tests.
Initially, the doctor will gather information regarding the patient's main complaints, associated symptoms, duration of symptoms, past medical history, and family medical history.
Following this, a thorough physical examination will be conducted, encompassing vital signs such as blood pressure, respiratory rate, pulse rate, and body temperature. The doctor will assess the patient's overall condition from head to toe.
Subsequently, diagnostic tests will be performed. These may include blood tests to assess hemoglobin levels and an Hb electrophoresis test to examine the shape of hemoglobin. These tests often aid in the diagnosis of hemoglobinopathy.
If necessary, a general practitioner may refer the patient to a hematologist a specialist in blood disorders, for further evaluation, confirmation of the diagnosis, and determination of the underlying cause.
Management
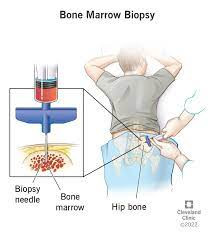
The management of hemoglobinopathy varies depending on its underlying cause. Treatment options may include iron supplementation, blood transfusions, and stem cell transplantation.
Complications
Complications of hemoglobinopathy may include:
- Acute Chest Syndrome
- Anemia
- Avascular Necrosis (Bone Tissue Death)
- Blood Clots
- Dactylitis (Hand-Foot Syndrome)
- Fever
- Infections
- Kidney Problems
Prevention
There is no specific method to prevent hemoglobinopathy since the disease is genetically inherited. However, suppose you are aware that you carry the hemoglobinopathy gene. In that case, seeking genetic counseling before planning to conceive can provide valuable insights into the risk of passing on the condition to your child. A genetic counselor can offer information about potential treatments, preventive measures, and reproductive options.
When to see a doctor?
If you experience the symptoms mentioned above and have a family history of hemoglobinopathy, it's essential to seek medical attention promptly.
Looking for more information about other diseases? Check here, yes!
- dr Hanifa Rahma
John Hopkins Medicine - G6PD (2021). Retrieved 13 April 2023, from https://www.hopkinsmedicine.org/health/conditions-and-diseases/g6pd-glucose6phosphate-dehydrogenase-deficiency#.
John Hopkins Medicine - Sickle Cell Disease (2021). Retrieved 13 April 2023, from https://www.hopkinsmedicine.org/health/conditions-and-diseases/sickle-cell-disease.
Mayo Clinic - Sickle Cell Disease (2022). Retrieved 10 April 2023, from https://www.mayoclinic.org/diseases-conditions/sickle-cell-anemia/symptoms-causes/syc-20355876.
Mayo Clinic - Thalassemia (2021). Retrieved 13 April 2023, from https://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/syc-20354995.
Taylor & Francis Online - Diagnostic Approach for Hemoglobinopathies (2019). Retrieved 10 April 2023, from https://www.tandfonline.com/doi/abs/10.1080/03630260701297071?journalCode=ihem20.
Thalassemia International Federation - Prevention and Diagnosis of Haemoglobinopathies (2019). Retrieved 13 April 2023, from https://thalassaemia.org.cy/wp-content/uploads/2019/11/Preventiondiagnosis-of-Hbpathies_BOOKLETNEW-1.pdf.