Definisi
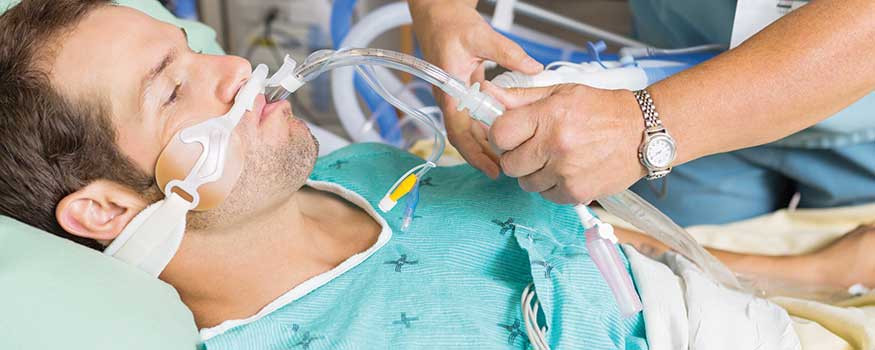
Fibrosis kistik adalah sebuah kondisi yang diwariskan dari orang tua ke anak, yang menyebabkan penumpukan lendir pada paru-paru dan saluran pencernaan. Jika kondisi ini tidak ditangani, dapat terjadi infeksi paru dan masalah pencernaan. Kondisi ini lebih banyak terjadi pada orang berkulit putih dibandingkan ras lainnya.
Penyebab
Fibrosis kistik adalah kondisi yang diturunkan secara genetik. Kondisi ini terjadi karena ada kesalahan pada gen CFTR yang mengatur pergerakan garam dan air pada sel. Akibatnya, terdapat lendir yang tebal dan lengket pada saluran napas, pencernaan, dan kelamin, serta peningkatan kadar garam pada keringat.
Orang yang memiliki gen yang menyebabkan kondisi ini tidak semuanya mengalami fibrosis kistik. Hal ini terjadi karena orang tersebut hanya memiliki satu gen yang menyebabkan kondisi tersebut. Untuk dapat menimbulkan gejala, gen bekerja secara berpasangan. Oleh karena itu, anak yang mengalami fibrosis kistik memiliki gen fibrosis kistik berpasangan, yang didapatkan dari masing-masing orang tua.
Faktor Risiko
Fibrosis kistik bersifat diwariskan, sehingga riwayat keluarga dengan kondisi serupa adalah faktor risiko terbesar. Meskipun fibrosis kistik terjadi pada semua ras, kondisi ini paling sering ditemukan pada keturunan Eropa Utara.
Gejala
Peningkatan jumlah dan kekentalan lendir pada saluran pernapasan dapat meningkatkan risiko masalah pernapasan, terutama infeksi. Selain itu, lendir ini dapat menyumbat saluran keluar enzim-enzim pencernaan yang berasal dari organ pankreas. Enzim-enzim ini sangat penting untuk memecah makanan agar dapat diserap, sehingga orang-orang dengan fibrosis kistik tidak dapat menyerap nutrisi dengan maksimal.
Gejala fibrosis kistik biasanya melibatkan sistem pernapasan dan sistem pencernaan. Gejala yang melibatkan sistem pernapasan adalah sebagai berikut:
- Batuk lama, menetap, dan berdahak
- Napas berbunyi
- Mudah lelah, tidak dapat berolahraga dalam waktu yang lama
- Infeksi paru berulang
- Pembengkakan dan peradangan rongga hidung, pilek
- Sinusitis (peradangan sinus) berulang
Sementara itu, gejala yang melibatkan sistem pencernaan adalah sebagai berikut:
- BAB berbau busuk dan berminyak
- Sulit mengalami kenaikan berat badan atau bertumbuh
- Sumbatan pada usus, terutama pada bayi yang baru lahir (disebut sebagai ileus mekonium)
- Sembelit yang parah dan berkepanjangan, termasuk mengedan yang terlalu keras ketika berusaha buang air, sehingga menyebabkan sebagian rektum (bagian usus yang mendekati lubang dubur) keluar dari lubang dubur, disebut sebagai prolaps rektal
Diagnosis
Fibrosis kistik dapat terdiagnosis mulai dari anak lahir. Pemeriksaan secara langsung dapat dilakukan untuk mencari adanya gejala-gejala yang mengarah ke fibrosis kistik, seperti pilek, batuk lama, kulit kuning, dan sebagainya. Selain itu, anak yang mengalami fibrosis kistik juga dapat memiliki kesulitan untuk bertumbuh selayaknya anak normal. Dokter juga dapat menanyakan riwayat keluarga dengan kondisi serupa untuk mengarahkan diagnosis. Selanjutnya, pemeriksaan laboratorium dapat dilakukan untuk mendiagnosis fibrosis kistik. Tes ini dapat melibatkan pengambilan keringat untuk mengukur kadar garam pada penderita, serta tes genetik untuk mencari gen yang menyebabkan fibrosis kistik.
Selain itu, pemeriksaan lainnya dapat dilakukan untuk mencari komplikasi fibrosis kistik pada tubuh penderita. Foto rontgen dapat dilakukan untuk mengetahui masalah pada paru serta usus pada bayi yang baru lahir. Computed tomography scan (CT scan) dapat dilakukan untuk mengetahui keparahan penyakit paru yang disebabkan oleh fibrosis kistik. Pemeriksaan lainnya dapat berupa pemeriksaan fungsi paru, untuk mengetahui seberapa parah penyakit paru mempengaruhi fungsi pernapasan. Selain itu, pemeriksaan dapat berupa immunoreactive trypsinogen (IRT) yang berfungsi untuk mengetahui masalah pencernaan pada bayi yang baru lahir.
Tata Laksana
Fibrosis kistik tidak dapat disembuhkan, namun dapat dikontrol. Jika kondisi ini tidak dikontrol, banyak komplikasi yang berpotensi merugikan hingga mengancam nyawa bagi penderita. Tata laksana biasanya bertujuan untuk mempertahankan fungsi paru, memastikan pertumbuhan berjalan normal, serta menangani masalah kesehatan yang muncul seiring dengan perjalanan penyakit.
Untuk mempertahankan fungsi paru, beberapa hal yang dapat dilakukan adalah meningkatkan pembersihan lendir dari saluran napas, terapi dengan obat-obatan hirup seperti obat asma, terapi fisik untuk dada dan mengalirkan lendir, menggunakan obat-obatan untuk mengencerkan dahak, serta penggunaan antibiotik untuk mencegah dan menangani infeksi paru segera.
Untuk memastikan pertumbuhan berjalan dengan normal, dokter dapat memberikan obat-obatan khusus untuk meningkatkan kadar enzim pencernaan dan multivitamin. Orang-orang dengan fibrosis kistik disarankan untuk mengonsumsi makanan tinggi lemak ditambah dengan konsumsi multivitamin yang larut dalam lemak. Tidak hanya itu, penderita juga disarankan untuk mengonsumsi kalori dalam jumlah tinggi untuk mempertahankan berat badan ideal. Jika penderita tinggal di daerah yang panas, konsumsi garam sangat dianjurkan untuk menggantikan garam yang hilang pada keringat.
Orang dengan fibrosis kistik boleh melakukan olahraga, bahkan sangat dianjurkan. Olahraga rutin dapat meningkatkan kekuatan fisik. Olahraga yang melibatkan badan bagian atas, seperti mendayung perahu kano, dapat memperkuat otot-otot pernapasan.
Komplikasi
Orang dengan fibrosis kistik memiliki risiko yang lebih tinggi untuk mengalami kondisi lainnya, seperti:
- Osteoporosis, atau tulang yang rapuh dan mudah patah
- Diabetes atau gula darah tinggi
- Infeksi sinus (rongga udara di antara tulang-tulang wajah) serta polip hidung atau benjolan bertangkai yang bersifat jinak pada rongga hidung
- Masalah pada organ hati
- Masalah kesuburan. Wanita dengan fibrosis kistik dapat memiliki anak, namun pria dengan fibrosis kistik kemungkinan besar tidak dapat memiliki anak tanpa bantuan dokter yang ahli dalam bidang ini
Orang dengan fibrosis kistik sebaiknya tidak bertemu tatap muka karena tingginya risiko menularkan infeksi serta tingginya risiko untuk terkena komplikasi apabila tertular infeksi.
Pencegahan
Fibrosis kistik cukup sulit dicegah karena sifatnya yang diwariskan. Jika Anda atau pasangan Anda berkerabat dekat dengan orang yang memiliki kondisi ini, sebaiknya Anda dan pasangan Anda melakukan tes genetik sebelum memutuskan untuk memiliki anak. Tes ini dilakukan dengan mengambil darah yang kemudian diperiksa di laboratorium dan dapat memperkirakan risiko Anda memiliki anak dengan fibrosis kistik. Apabila Anda sedang hamil dan tes genetik menunjukkan bahwa janin memiliki risiko fibrosis kistik, dokter dapat melakukan beberapa tes lainnya untuk janin Anda.
Tes genetik tidak wajib bagi semua orang. Sebelum Anda memutuskan untuk dites, Anda dapat berkonsultasi kepada dokter atau konselor genetik mengenai efek psikologis dari hasil tes yang akan dilakukan.
Kapan harus ke dokter?
Jika Anda atau anak Anda memiliki gejala fibrosis kistik, Anda dapat berkonsultasi kepada dokter untuk melakukan tes diagnosis kondisi tersebut. Fibrosis kistik memerlukan kontrol yang rutin dan teratur, minimal setiap 3 bulan sekali. Anda dapat berkonsultasi kepada dokter jika mengalami gejala baru atau memburuk, seperti jumlah lendir yang lebih banyak daripada biasanya, perubahan warna lendir, rasa lelah terus-menerus, penurunan berat badan yang tidak diinginkan, atau sembelit yang parah.
Anda sebaiknya segera ke IGD terdekat jika batuk berdarah, nyeri dada, sesak napas, atau mengalami nyeri perut yang luar biasa.
- dr Anita Larasati Priyono
Cystic fibrosis. (2021). Retrieved 2 March 2022, from https://www.nhs.uk/conditions/cystic-fibrosis/
Cystic fibrosis - Symptoms and causes. (2021). Retrieved 2 March 2022, from https://www.mayoclinic.org/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700
Sharma, G. (2020). Cystic Fibrosis: Practice Essentials, Background, Pathophysiology. Retrieved 2 March 2022, from https://emedicine.medscape.com/article/1001602-overview
Yu, E., & Sharma, S. (2021). Cystic Fibrosis. Retrieved 2 March 2022, from https://www.ncbi.nlm.nih.gov/books/NBK493206/